RKI Initial Validation Autoclave
 RKI 1: Legal Requirements & Overview
RKI 1: Legal Requirements & Overview  RKI 2: Proper Pre-Cleaning
RKI 2: Proper Pre-Cleaning  RKI 3: Cleaning & Disinfection
RKI 3: Cleaning & Disinfection 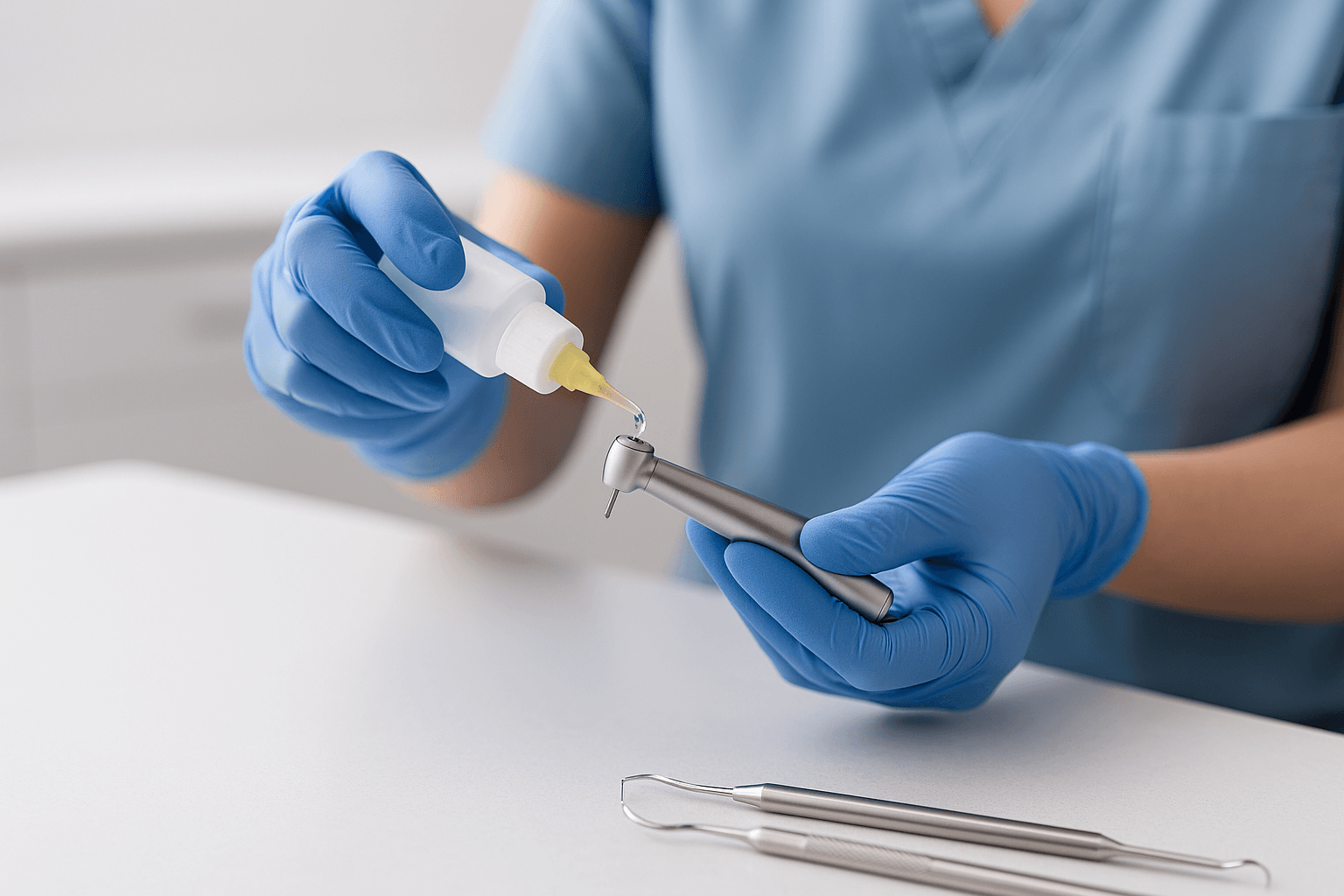 RKI 4: Instrument Care
RKI 4: Instrument Care 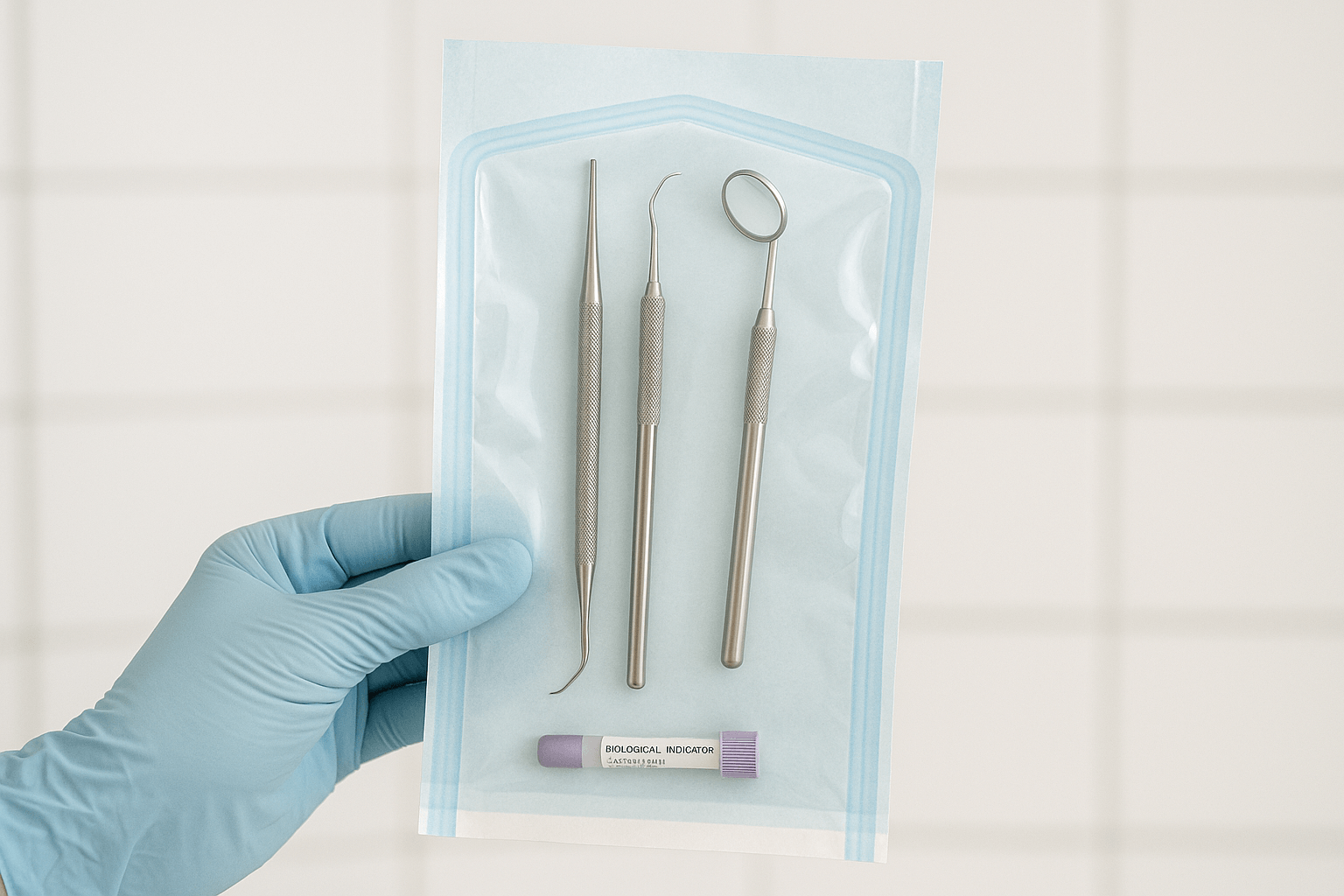 RKI 5: Instrument Packaging
RKI 5: Instrument Packaging  RKI 6: Sterilisation in the Autoclave
RKI 6: Sterilisation in the Autoclave  RKI 7: Release & Documentation
RKI 7: Release & Documentation  RKI 8: Storage of Sterile Instruments
RKI 8: Storage of Sterile Instruments 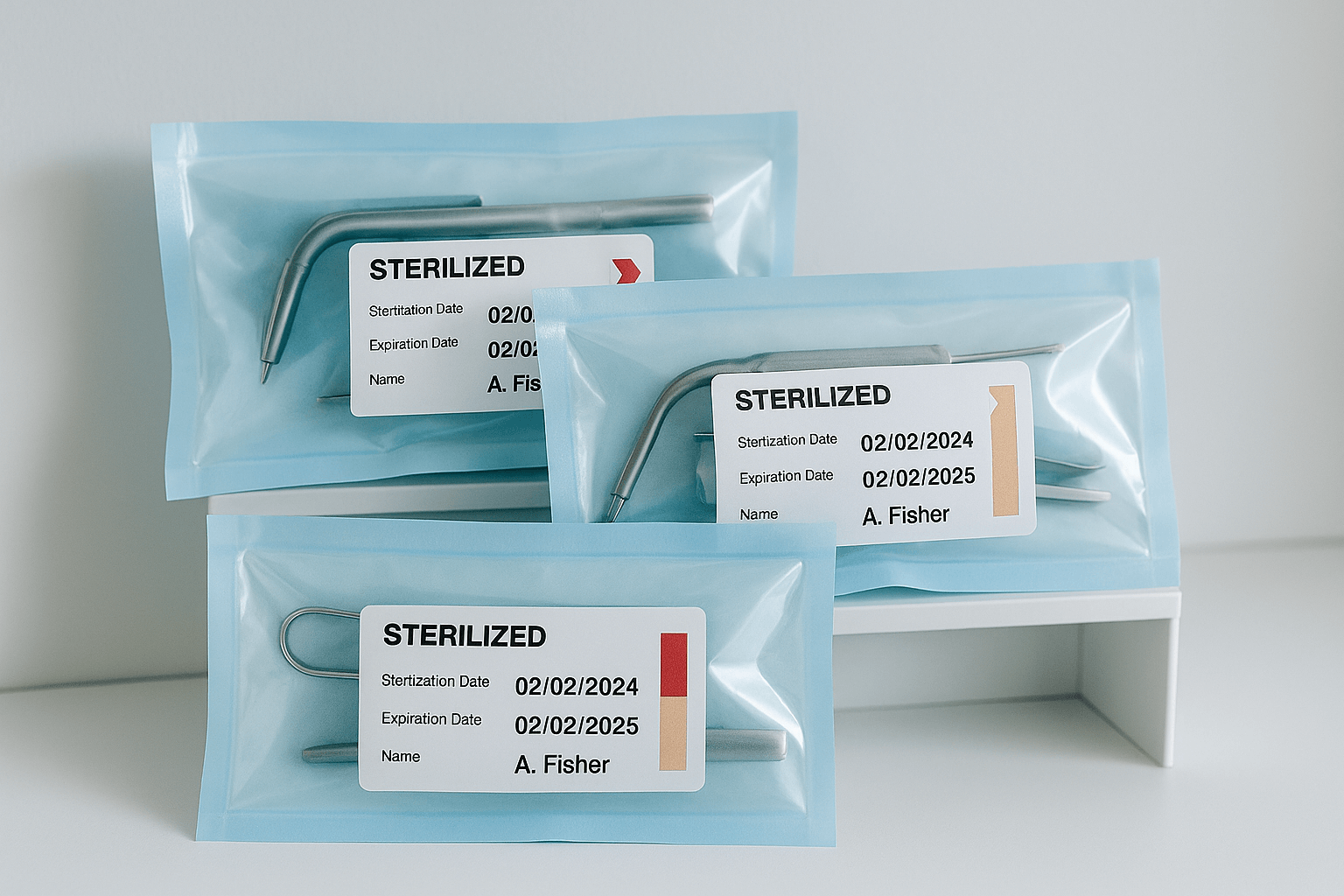 RKI 9: Labelling After Sterilisation
RKI 9: Labelling After Sterilisation 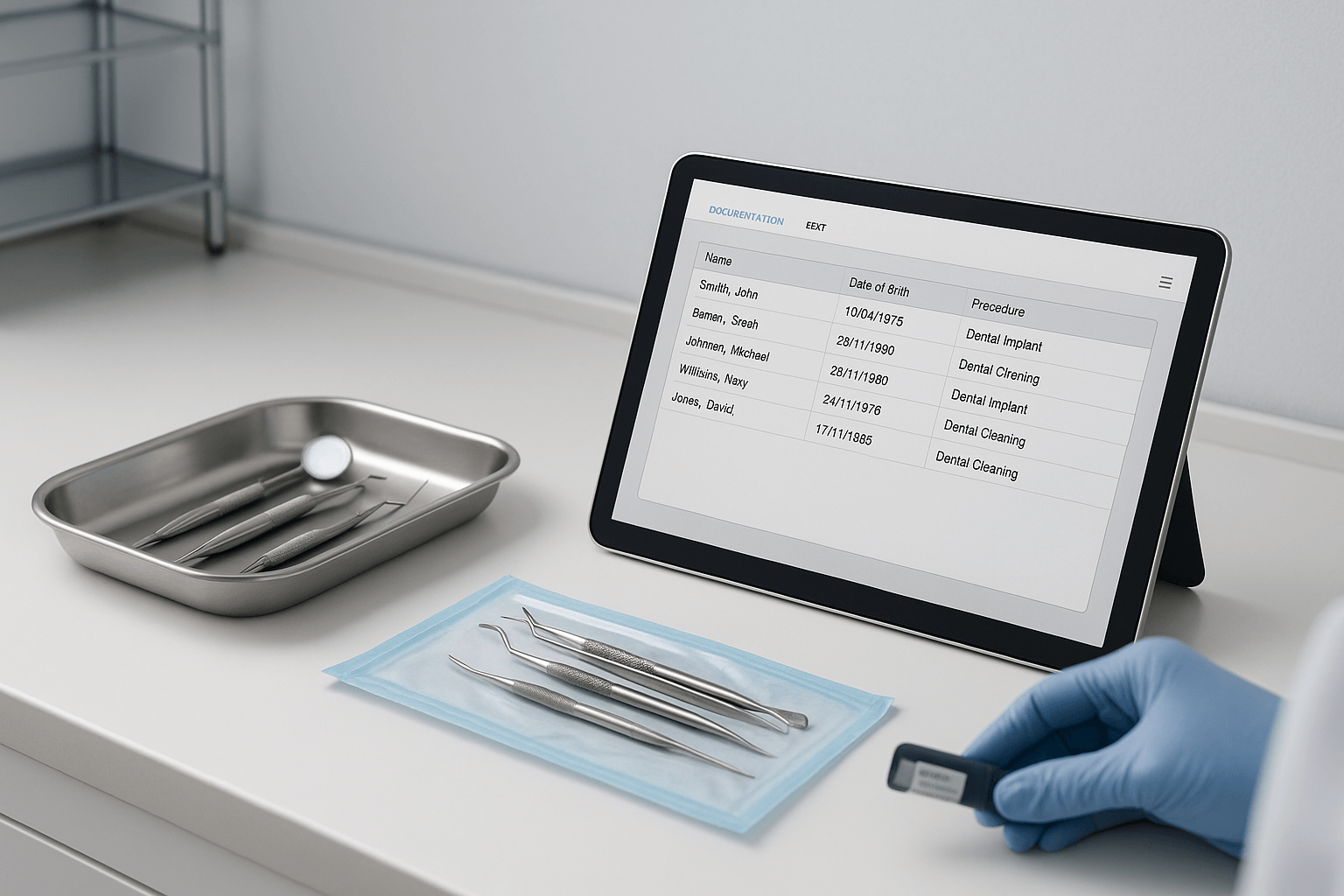 RKI 10: Documentation After Patient Use
RKI 10: Documentation After Patient Use  RKI 11: Hygiene Room per KRINKO
RKI 11: Hygiene Room per KRINKO  RKI 12: Initial Validation Class B Autoclave
RKI 12: Initial Validation Class B Autoclave
Meet RKI requirements – free of charge
Use the free ClavioSoft to run an audit check of your practice in just a few minutes – the software automatically generates any missing documentation. This saves you up to 90 % of the work.

Initial Validation of a Class B Autoclave: IQ, OQ & PQ Step by Step
Validation demonstrates that your Class B autoclave sterilises reproducibly and safely. Under MPBetreibV § 8, reprocessing must be carried out using suitable, validated procedures. The (initial) validation is divided into IQ, OQ and PQ based on DIN EN ISO 17665 and DIN SPEC 58929 as well as KRINKO Annex 4; DIN EN 13060 is the underlying equipment/type-testing standard (process type B).
1Preparation & Planning
· Purpose and scope of the validation to be defined (initial validation after installation).
· Document review: risk assessments, standard operating procedures, training records.
· Standards compliance: verification against DIN EN 13060 and DIN EN ISO 17665.
2Installation Qualification (IQ)
Verifies that the equipment has been correctly delivered, installed and commissioned:
· Environment: suitable location, room temperature, humidity. · Equipment condition: integrity and completeness of accessories. · Utilities: power supply and, where applicable, water connection. · Initial tests: air leakage test (vacuum/leak test) and empty-chamber test.
3Operational Qualification (OQ)
Ensures that the autoclave operates within the specified parameters:
· Process sequence: testing with a defined worst-case load. · Air removal & steam penetration: using a suitable process challenge device (PCD for hollow instruments, e.g. Helix test). · Fault detection: verification of the fault-detection system per manufacturer instructions, documented in the qualification report.
4Performance Qualification (PQ)
The most important step – demonstrating sterilisation performance with real loads:
· Reference loads: multiple cycles with different, practice-typical loads. · Data recording: measurement of pressure and temperature profiles at the critical points of the test load using an independent, calibrated measurement system (data logger); microbiological testing where physical measurement is not possible. · Drying test: residual moisture on the sterilised goods determined by weight difference before/after the cycle, within the specified limits.
5Routine Checks & Final Report
Routine checks ensure long-term stability: visual inspection of door seals and loading trays, filter replacement per manufacturer instructions, regular tests. A final report then summarises all results, conditions and recommendations (including the next revalidation date) – a key document to present during a practice inspection. Ongoing batch documentation and full traceability are conveniently managed with the free ClavioSoft.
Find the right Class B autoclave Order validation service
Important Additional Points per RKI & Standards
These points are frequently overlooked in practice but are essential for correct and legally compliant implementation:
- Definition and justification of the worst-case load: Before PQ, the operator together with the validator defines all practice-relevant load configurations and identifies the "most challenging" (worst-case) load with a documented, traceable justification. Only these validated configurations may subsequently be used in routine operation; an equivalence demonstration must be provided for actual loads.Source: KRINKO/BfArM 2012, Annex 4: "Description of all load configurations including the most challenging load(s) (with corresponding justification)"; PQ "Definition and documentation of test loads including equivalence demonstration"
- Risk classification of medical devices to be reprocessed as a prerequisite: The basis of any validation is the classification of the medical devices to be reprocessed (non-critical / semi-critical / critical per RKI/BfArM). This risk classification determines loads, scope of testing and required process challenge devices; it must be established as an operator document before PQ.Source: KRINKO/BfArM 2012, Annex 4: "Risk classification of medical devices to be sterilised per RKI-BfArM recommendation"; Table 1 (classification)
- Feed-water quality as an IQ checkpoint: During IQ, the quality of the feed water for the Class B autoclave must be tested and documented against the requirements of DIN EN 13060, Annex C. Unsuitable feed water causes corrosion, deposits and faulty cycles and jeopardises sterilisation performance.Source: KRINKO/BfArM 2012, Annex 4: "Feed-water quality: DIN EN 13060, Annex C"
- Packaging and sealing process as part of the initial validation: If wrapped sterilised goods are reprocessed, the sterile barrier system and sealing process form part of the validated overall process. For heat sealers, the critical parameters are temperature and contact pressure; the seal width must be at least 6 mm and the minimum distance between the seal and the medical device 3 cm. Packaging must comply with DIN EN 868-2 ff. and DIN EN ISO 11607-1; routine checks include ink/seal-check tests and peel-strength testing.Source: KRINKO/BfArM 2012, Annex 4, "Sterile-goods packaging" and "Heat sealers"; DIN EN ISO 11607-1; DIN EN 868-2
- Selection of routine process challenge devices (PCD) and chemical indicators as a validation output: A key outcome of validation is the definition of the routine checks required: selection of suitable process challenge devices (PCD), where hollow instruments are sterilised (per DIN EN 13060 Annex A), and selection of chemical indicators and the process assessment system. These specifications are documented in the validation report and govern subsequent batch release.Source: KRINKO/BfArM 2012, Annex 4, "Specifications for routine checks during operation"; DIN EN 13060 Annex A/B
- Operator training and designation of authorised release personnel (authorisation list): Initial validation and commissioning include documented training of operating personnel in operation and fault response, as well as the creation of an authorisation list of persons permitted to release batches (with proof of qualification and defined release criteria).Source: KRINKO/BfArM 2012, Annex 4: IQ "Training in operation ... and fault response", "Regular instruction of ... personnel", Release "Authorisation list" and "Release criteria"
- Content and retention of the validation report: In addition to results, the validation report must include recorder printouts/logger protocols, photographic documentation of the tested loads, specifications for routine checks and the due date for the next periodic re-qualification; it must be archived in an audit-proof manner. This tamper-proof filing and retrieval of protocols is handled in just a few clicks with the free ClavioSoft.Source: KRINKO/BfArM 2012, Annex 4: "Validation report with recorder printouts and photographic documentation"; "Determination of the due date for periodic re-qualifications ... validation report"
- Prior cleaning/disinfection as a validation prerequisite: Sterilisation processes can only be fully validated on the assumption that they are applied to residue-free cleaned medical devices. Initial validation of the autoclave therefore requires a standardised, upstream cleaning and disinfection process.Source: KRINKO/BfArM 2012, Section 1.3: "Sterilisation processes can be fully validated on the assumption of their application to residue-free cleaned medical devices"
Required Equipment, Consumables & Accessories
Checklist for this step – items you should have ready (shopping list):
Equipment
- Class B autoclave (process type B per DIN EN 13060), installed and ready for operation with complete manufacturer documentation Order in our shop
- Independent, calibrated measurement/data-logging system for temperature and pressure (traceably calibrated, with valid calibration certificate)
- Suitable process challenge device (PCD) for hollow instruments (e.g. Helix test) per DIN EN 13060 Annex A Order in our shop
- Measuring equipment or built-in programme for air-leakage/vacuum test and empty-chamber test
- Precision balance for determining drying/residual moisture of the sterilised goods (weight difference before/after cycle)
- Conductivity meter for testing feed-water quality (DIN EN 13060 Annex C)
- Sealing device (if wrapped goods are reprocessed) for testing seal width and seal strength Order in our shop
- Digital documentation software for protocol and report recording (optional) Order in our shop
Consumables
- Practice-typical reference/test loads including the defined worst-case load (own, cleaned instruments)
- Test packs/indicators for steam-penetration testing in the PCD Order in our shop
- Chemical indicators of the required classes (process indicator Class 1 and indicators suitable for the test) Order in our shop
- Biological indicators where applicable for microbiological testing at points where physical measurement is not possible
- Sterile barrier systems/packaging materials per DIN EN 868-2 ff. / DIN EN ISO 11607-1 (for wrapped reprocessing) Order in our shop
- Seal-check/ink-test strips for seal-seam inspection (for wrapped reprocessing) Order in our shop
- Treated feed water of suitable quality (e.g. demineralised) for test cycles Order in our shop
- Replacement seal and cleaning agent per manufacturer instructions (for IQ visual inspection)
Accessories
- Loading trays/racks and holders matching the unit Order in our shop
- Sensor positioning aids for placing loggers at the critical points of the load
- Calibration certificates/traceability records for the measuring instruments used
- Equipment recorder/printer or interface for protocol output (recorder printouts) Order in our shop
- Camera for the required photographic documentation of load configurations
- Manufacturer documentation: equipment, operating and maintenance instructions, type-test/CE certificate
- Documentation templates: risk classification, load lists, IQ/OQ/PQ protocols, installation/handover protocol, authorisation/release list, training record, validation report template Create digitally with ClavioSoft
Templates & References
- KRINKO/BfArM 2012, Bundesgesundheitsbl. 55:1244–1310 – Section 1.3; Annex 4 (small sterilisers).
- DIN EN 13060 (steam small sterilisers); DIN EN ISO 17665 (steam sterilisation, validation).
- MPBetreibV § 8 (validated procedures).
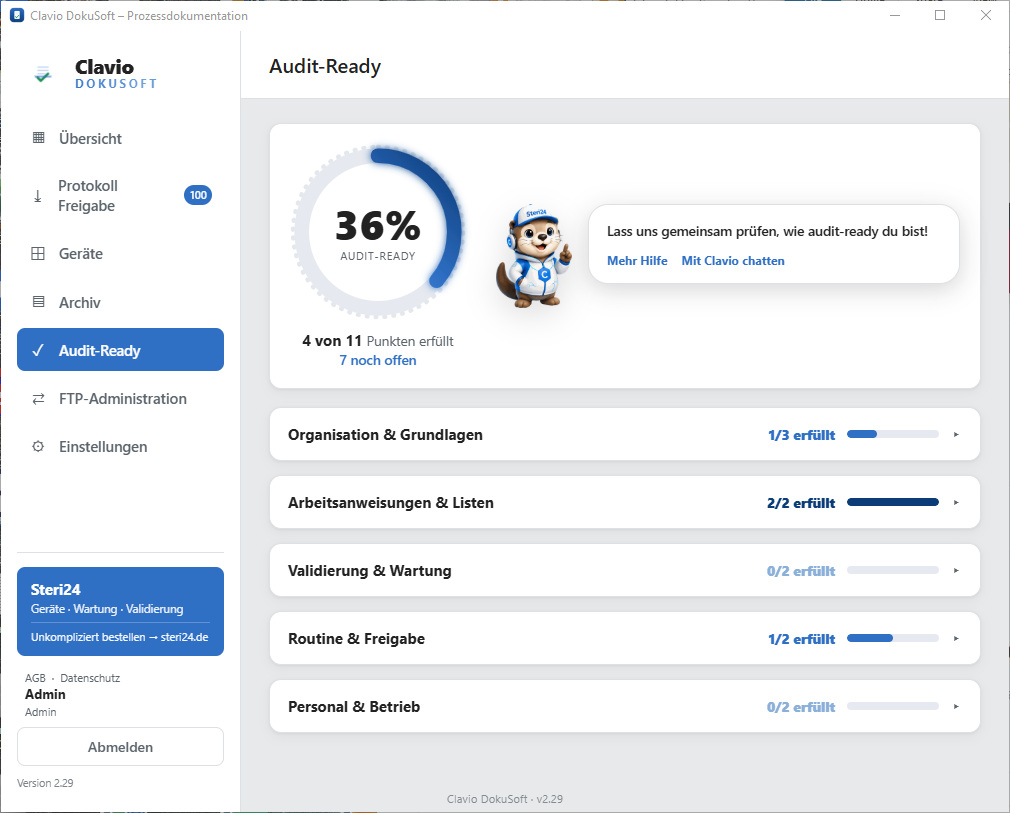
How audit-ready is your practice?
Find out in just a few minutes how well you are prepared for the next inspection — category by category, with clear progress tracking and specific open items. Completely free with ClavioSoft.
.png "Preview: Clavio DokuSoft")



Frequently Asked Questions
RKI Initial Validation Autoclave – key questions answered clearly.