RKI Legal Requirements
 RKI 1: Legal Requirements & Overview
RKI 1: Legal Requirements & Overview  RKI 2: Proper Pre-Cleaning
RKI 2: Proper Pre-Cleaning  RKI 3: Cleaning & Disinfection
RKI 3: Cleaning & Disinfection  RKI 4: Instrument Care
RKI 4: Instrument Care  RKI 5: Instrument Packaging
RKI 5: Instrument Packaging  RKI 6: Sterilization in the Autoclave
RKI 6: Sterilization in the Autoclave  RKI 7: Release & Documentation
RKI 7: Release & Documentation  RKI 8: Storage of Sterile Instruments
RKI 8: Storage of Sterile Instruments 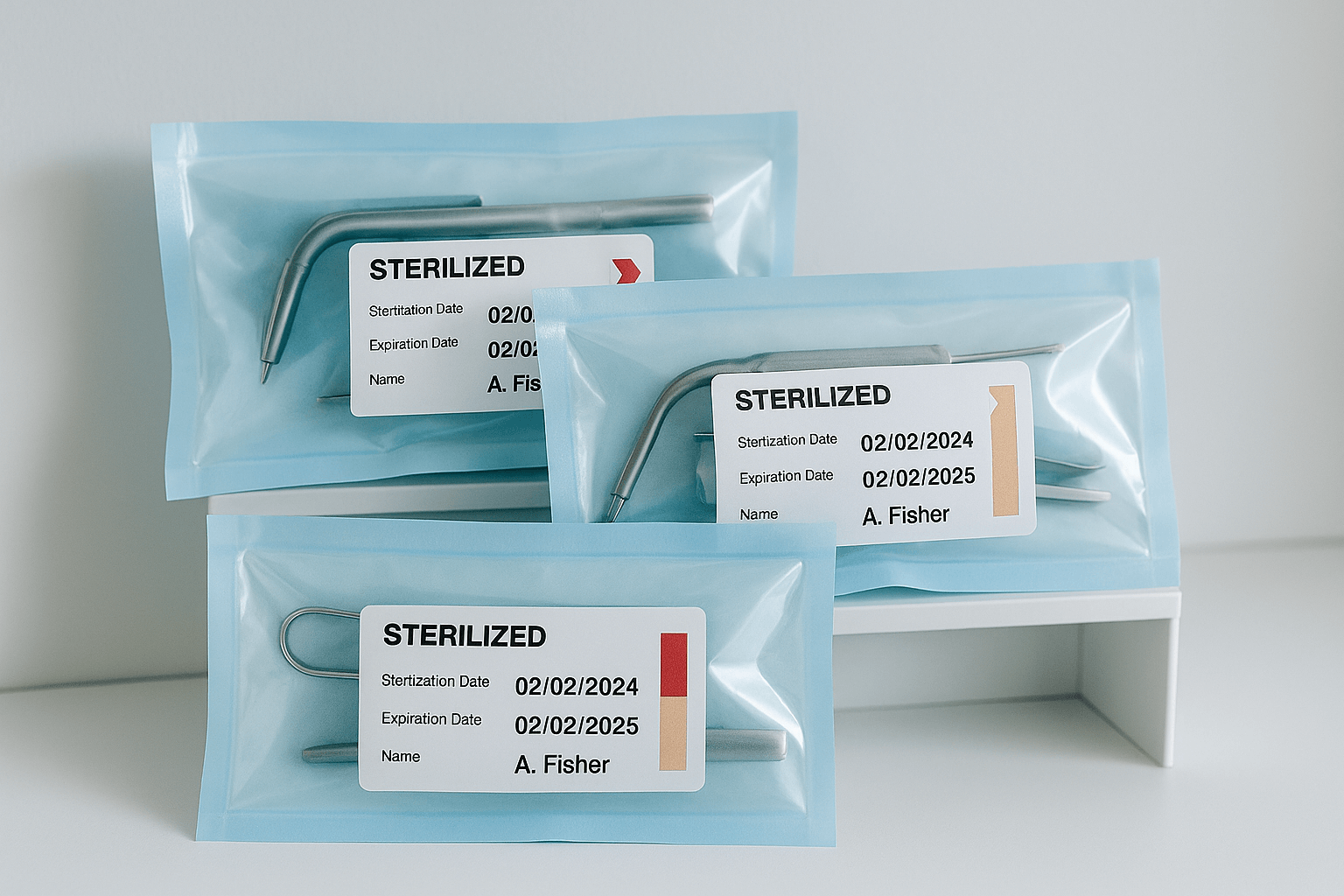 RKI 9: Labelling After Sterilization
RKI 9: Labelling After Sterilization  RKI 10: Documentation After Patient Use
RKI 10: Documentation After Patient Use  RKI 11: Hygiene Room per KRINKO
RKI 11: Hygiene Room per KRINKO  RKI 12: Initial Validation Klasse B Autoclave
RKI 12: Initial Validation Klasse B Autoclave
Meet RKI requirements – free of charge
Use the free ClavioSoft to run an audit check of your practice in just a few minutes – the software automatically generates any missing documentation. This saves you up to 90 % of the work.

Instrument Reprocessing per RKI: Legal Requirements & Risk Classes
In Germany, the reprocessing of reusable medical devices is legally mandatory and clearly regulated. Anyone who uses instruments more than once must reprocess them using a validated procedure, follow the KRINKO recommendation issued by the Robert Koch Institute (RKI) and document every step in a verifiable manner. Responsibility always lies with the practice owner (§ 8 MPBetreibV).
This page is the starting point for our 12-part step-by-step guide to RKI-compliant instrument reprocessing. It explains – even without prior knowledge – which laws apply, how to assign each of your instruments to the correct risk class and which reprocessing procedure is mandatory as a result. By the end, you will know exactly what needs to be done in your practice.
1What does the law require?
Four regulatory frameworks interlock. You do not need to know the laws by heart – the important thing is to understand what they amount to:
| Regulation | What it covers | What this means for you |
|---|---|---|
| IfSG Infection Protection Act | Fundamental protection against infections in healthcare facilities. | You must actively prevent the transmission of pathogens – clean instruments are mandatory. |
| MDR & MPDG EU Medical Device Regulation 2017/745 + Implementation Act | Replaced the former Medical Devices Act (MPG) in 2021 and governs the safe handling of medical devices. | Reusable devices may only be reprocessed in a way that preserves their safety and function. |
| MPBetreibV Medical Device Operator Ordinance, § 8 | Specifies how reprocessing must be carried out – and expressly refers to the KRINKO/RKI recommendation. | The central compliance manual for your practice (see below). |
| KRINKO/RKI Recommendation "Requirements for hygiene in the reprocessing of medical devices" | Describes the actual reprocessing procedure step by step. | Your concrete working instructions – these are exactly what we implement across the 12 chapters. |
The core obligations under § 8 MPBetreibV
The decisive paragraph is § 8 of the MPBetreibV. It specifically requires you as the operator to:
1) Follow manufacturer instructions – reprocess every instrument as approved by the manufacturer (cf. DIN EN ISO 17664).
2) Validated procedure – cleaning, disinfection and sterilization must be demonstrably effective and reproducible.
3) Obligation to provide evidence – the success of each reprocessing cycle must be documented (batch documentation). This batch and release documentation can be completed in just a few clicks with the free ClavioSoft – including audit-proof archiving.
4) Follow the KRINKO/RKI recommendation – it represents the acknowledged state of science and technology.
5) Qualified personnel – anyone who reprocesses instruments requires the necessary expertise (training or qualification; cf. KRINKO Annex 6).
2How to proceed in practice – in 4 steps
Before you reprocess a single instrument, you must define in writing whether, with what and under what conditions each product is to be reprocessed. The KRINKO explicitly requires this – this determination is also a central component of your hygiene plan and easier to implement than it sounds:
Source: KRINKO/BfArM Recommendation 2012, Sect. 1.2.1 "Risk assessment and classification".Step 1: List all reusable instruments
Start by creating a complete list of all products you intend to reuse – without any evaluation, simply list everything. For example:
| Name | Quantity (example) |
|---|---|
| ECG electrodes | 12 |
| Ear speculum | 5 |
| Ear irrigation syringe | 2 |
| Speculum | 3 |
| Flexible endoscopes (e.g. gastroscope) | 2 |
| Scissors | 5 |
| Surgical forceps | 5 |
| Anatomical forceps | 5 |
| Wound retractor | 5 |
| Pean clamp | 5 |
| Curette | 5 |
| Needle holder | 5 |
| MIC trocar | 4 |
| ERCP catheter | 2 |
Step 2: Assign the risk class to each instrument
The risk class depends on what the instrument comes into contact with on the patient. The KRINKO distinguishes three main classes:
| Contact with the patient | Risk class | Sub-classification |
|---|---|---|
| Contact with intact skin only | Non-critical | – |
| Contact with mucous membranes or pathologically altered skin | Semi-critical (A or B) | A: without areas difficult to access for steam |
| B: with lumens / areas difficult to access | ||
| Penetrates skin/mucous membranes and comes into contact with blood, internal tissues or organs (also: application of blood/sterile medicinal products; contact with urine inside the body) | Critical (A, B or C) | A: without areas difficult to access for steam |
| B: with lumens / areas difficult to access | ||
| C: particularly high requirements per the manufacturer (e.g. not steam-sterilizable) |
Here is how the classification looks using an example:
| Name | Quantity (example) | Risk classification per RKI |
|---|---|---|
| ECG electrodes | 12 | Non-critical |
| Ear speculum | 5 | Semi-critical A |
| Ear irrigation syringe | 2 | Semi-critical A |
| Speculum | 3 | Semi-critical A |
| Flexible endoscopes (e.g. gastroscope) | 2 | Semi-critical B |
| Scissors | 5 | Critical A |
| Surgical forceps | 5 | Critical A |
| Anatomical forceps | 5 | Critical A |
| Wound retractor | 5 | Critical A |
| Pean clamp | 5 | Critical A |
| Curette | 5 | Critical A |
| Needle holder | 5 | Critical A |
| MIC trocar | 4 | Critical B |
| ERCP catheter | 2 | Critical C |
Find the right Class B autoclave
Step 3: Determine the correct procedure for each risk class
The risk class determines which reprocessing steps are mandatory – from pre-cleaning to sterilization. This table translates each class into specific actions:
| Risk class | Reprocessing |
|---|---|
| Non-critical | Pre-cleaning: no · Cleaning and disinfection: yes (manual or automated) with a proven effective disinfectant or method; automated processing is preferred due to better reproducibility · Packaging: no · Sterilization: no · Labelling: no |
| Semi-critical A | Pre-cleaning: optional · Cleaning followed by effective disinfection with a proven spectrum of activity (bactericidal including mycobacteria, fungicidal, virucidal) – manual or automated. An ultrasonic cleaner only supports cleaning and does not replace disinfection; a washer-disinfector (WD) performs cleaning and disinfection in a single step. · Packaging: no · Sterilization: optional only · Labelling: no |
| Semi-critical B | Pre-cleaning: yes, immediately after use (e.g. immersion) · Cleaning and disinfection preferably automated/thermal in a washer-disinfector (WD); ultrasonic cleaning is at best a preliminary cleaning step and not a disinfection method. Manual processing only if automated processing is not possible · Packaging: no · Sterilization: mandatory when used in a sterile body cavity · Labelling: no |
| Critical A | Pre-cleaning: optional · Cleaning and disinfection: preferably automated in a washer-disinfector (WD); an ultrasonic cleaner serves only as a supporting cleaning step (e.g. pre-cleaning) and does not replace disinfection · Packaging: yes · Sterilization: yes, wrapped · Labelling: yes |
| Critical B | Pre-cleaning: yes, immediately after use · Cleaning + disinfection: yes, in a washer-disinfector · Packaging: yes · Sterilization: yes, wrapped (Klasse B) · Labelling: yes |
| Critical C | Pre-cleaning / cleaning + disinfection / sterilization: each according to the instrument manufacturer's instructions · Packaging: yes · Labelling: yes |
Step 4: Combine everything into a comprehensive overview
Finally, merge both tables into one overview. It shows at a glance which equipment and steps are required for each instrument – and simultaneously serves as your documented specification in accordance with the KRINKO:
| Name | Quantity | Risk classification | Pre-cleaning | Cleaning + disinfection | Labelling | Sterilization |
|---|---|---|---|---|---|---|
| ECG electrodes | 12 | Non-critical | No | Yes | No | No |
| Ear speculum | 5 | Semi-critical A | Optional | Yes | No | Optional |
| Ear irrigation syringe | 2 | Semi-critical A | Optional | Yes | No | Optional |
| Speculum | 3 | Semi-critical A | Optional | Yes | No | Optional |
| Flexible endoscopes (e.g. gastroscope) | 2 | Semi-critical B | Yes | Yes | No | When used in a sterile body cavity |
| Scissors | 5 | Critical A | Optional | Yes | No | Yes |
| Surgical forceps | 5 | Critical A | Optional | Yes | No | Yes |
| Anatomical forceps | 5 | Critical A | Optional | Yes | No | Yes |
| Wound retractor | 5 | Critical A | Optional | Yes | No | Yes |
| Pean clamp | 5 | Critical A | Optional | Yes | No | Yes |
| Curette | 5 | Critical A | Optional | Yes | No | Yes |
| Needle holder | 5 | Critical A | Optional | Yes | No | Yes |
| MIC trocar | 4 | Critical B | Yes | Yes | Optional | Yes |
| ERCP catheter | 2 | Critical C | Yes | Yes | Yes | Yes |
3Templates & documents for download
These templates and original sources help you implement the steps described above directly – ready to print and adapt to your practice:
Important additional points per RKI & standards
These points are frequently overlooked in practice but are important for correct and legally compliant implementation:
- Extension of the referenced standards per process step: In addition to DIN EN 13060 and DIN EN ISO 17664, supplement with the standards relevant to each individual step: DIN EN ISO 15883 (washer-disinfectors/thermal disinfection), DIN EN ISO 17665 (validation of steam sterilization) and DIN EN ISO 11607-1/-2 and DIN 58953 (sterile barrier systems/packaging). These standards are listed as applicable in the standards annex of the KRINKO/BfArM Recommendation 2012.Source: KRINKO/BfArM 2012, Standards Annex (lists DIN EN ISO 15883, 17665, 11607, DIN 58953)
- Pre-treatment/collection at the point of use: Include proper pre-treatment at the point of use as the first step: remove gross contamination immediately after use, prevent drying of blood/tissue (e.g. wiping, flushing working channels, defining disposal timeframes) and avoid fixating procedures before cleaning. The KRINKO explicitly counts pre-treatment, collection and pre-cleaning as the first working step of reprocessing.Source: KRINKO/BfArM 2012, Sect. 2.2.1 'Preparation of reprocessing (pre-treatment, collection, pre-cleaning ...)'
- Release for use as a separate, documented step: Include the documented release for use as the conclusion of reprocessing: comparison of recorded process parameters with validation reports, performance and documentation of routine tests, inspection of the packaging for integrity and dryness as well as labelling; the persons authorized to release must be designated in writing. Only then may the sterile goods be used. The associated release documentation can be managed traceably and paperlessly with the free ClavioSoft.Source: KRINKO/BfArM 2012, Sect. 2.2.7 'Release for use'; MPBetreibV § 8
- Specify validation of procedures (initial and re-validation): The article mentions 'validated procedure' but does not explain its implementation: cleaning/disinfection and sterilization processes must be validated before first use and re-tested at regular intervals; changes to loading, programme or packaging require a renewed assessment. Specific test intervals are determined by the manufacturer and/or standard requirements (do not state a blanket interval).Source: KRINKO/BfArM 2012, Sect. 1.3 'Validation'; Standards Annex (DIN EN ISO 17665, DIN EN ISO 15883)
- Daily routine tests and indicators in sterilization operations: Include the ongoing routine tests for the Class B autoclave: vacuum/leak test and steam penetration test using a suitable test device (hollow device) as well as batch control with process indicators. These tests are a prerequisite for batch release.Source: DIN EN 13060 (Class B sterilizer tests, Annex B); KRINKO/BfArM 2012, Sect. 2.2.7
- Water quality for steam sterilization and automated reprocessing: Include the water quality requirement: the small steam sterilizer requires feed water in accordance with DIN EN 13060 (Annex C); treated water is also needed for the final rinse in the washer-disinfector to prevent staining, corrosion and equipment damage.Source: KRINKO/BfArM 2012, Annex 4 'Feed water quality: DIN EN 13060, Annex C'
- Structural/zonal separation of clean and soiled areas and personnel protection: Include the KRINKO requirement for separated work areas (soiled: receiving/cleaning – clean: packaging/release – storage) as well as personal protective equipment in the soiled area (suitable gloves, protective eyewear, protective clothing) to prevent cross-contamination and protect personnel.Source: KRINKO/BfArM 2012, Annex 5 (zone/area separation soiled-clean-storage) and Sect. 2.2.1 (occupational safety, TRBA 250)
- Verification of cleaning effectiveness (cleanliness/residual protein): Include the verification of cleaning effectiveness: visual/magnification inspection for cleanliness and integrity; for non-automated (manual) procedures the cleaning effectiveness must be separately verified (e.g. spot-check protein determination; the KRINKO specifies a warning value of 100 µg protein per instrument).Source: KRINKO/BfArM 2012, Sect. 2.2.2 (inspection for cleanliness; warning value 100 µg protein)
- Risk-class-specific expertise requirements: Specify personnel requirements in detail: for 'critical B' the KRINKO additionally requires proof of recognised training for the person carrying out the reprocessing; for 'critical C' a certification of the quality management system (DIN EN ISO 13485 by a recognised body) is also required.Source: KRINKO/BfArM 2012, Table 1 (footnotes to critical B/C) and Annex 6 'Personnel expertise'; DIN EN ISO 13485
Required equipment, consumables & accessories
Checklist for this work step – what you should have ready (shopping list):
Equipment
- Class B small steam sterilizer (autoclave) per DIN EN 13060 for the wrapped sterilization of critical instruments Order here in the shop
- Washer-disinfector (WD) per DIN EN ISO 15883 for preferred automated cleaning and disinfection Order here in the shop
- Ultrasonic cleaner as a supporting cleaning/pre-cleaning device (does not replace disinfection) Order here in the shop
- Water treatment/demineralization system for demineralized feed and rinse water Order here in the shop
- Sealing device for transparent sterilization packaging (validatable, per DIN EN ISO 11607-2) Order here in the shop
- Digital documentation software or printer/data logger for batch and release documentation Order here in the shop Create digitally with ClavioSoft
Consumables
- Cleaning and disinfection agents with a proven spectrum of activity (bactericidal incl. mycobacteria, fungicidal, virucidal) Order here in the shop
- Transparent sterilization packaging/sterile barrier system and nonwoven/crepe per DIN EN ISO 11607-1 / DIN 58953 Order here in the shop
- Process/chemical indicators and indicator tape for batch control Order here in the shop
- Test device/test system for vacuum and steam penetration testing (hollow device test piece) Order here in the shop
- Demineralized/distilled water as feed and final rinse water Order here in the shop
- Care/lubricant for hinged instruments (sterilizable, manufacturer-approved) Order here in the shop
- Residual protein test/cleaning indicators for verification of cleaning effectiveness (especially for manual reprocessing)
- Personal protective equipment: fluid-resistant disposable gloves, protective eyewear, protective clothing
Accessories
- Writable labels/pen or label printer for content, batch and expiry labelling Order here in the shop Create digitally with ClavioSoft
- Instrument trays, cassettes and storage aids matching the validated reference load Order here in the shop
- Sorting/transport containers with lids for collection and transport (clean/soiled separated) Order here in the shop
- Templates/forms: risk assessment list, standard operating procedure 'critical B', sample tray list, loading pattern, batch release record Create digitally with ClavioSoft
- Magnifying glass or illuminated magnifier for visual inspection of cleanliness and integrity
- Separate and labelled work surfaces for soiled and clean areas
- Current manufacturer reprocessing instructions (DIN EN ISO 17664) for each instrument as an accessible reference
Sources & legal basis
- Infection Protection Act (IfSG).
- Regulation (EU) 2017/745 on medical devices (MDR) and Medical Device Regulation Implementation Act (MPDG).
- Medical Device Operator Ordinance (MPBetreibV), in particular § 8 "Reprocessing of medical devices" (presumption effect para. 2).
- KRINKO/BfArM: "Requirements for hygiene in the reprocessing of medical devices", Bundesgesundheitsbl. 2012; 55:1244–1310 – in particular Sect. 1.2.1 (risk assessment & classification) and 2.2 (implementation); Annex 6 (personnel expertise).
- DIN EN ISO 17664 (manufacturer reprocessing instructions); DIN EN 13060 (small steam sterilizers, Klasse B).
This article summarises the cited legal and standards sources in a practice-oriented manner and does not replace the study of the original documents. In case of doubt, the respectively current versions of the laws, ordinances and recommendations apply.
How audit-ready is your practice?
Find out in just a few minutes how well you are prepared for the next inspection — category by category, with clear progress tracking and specific open items. Completely free with ClavioSoft.
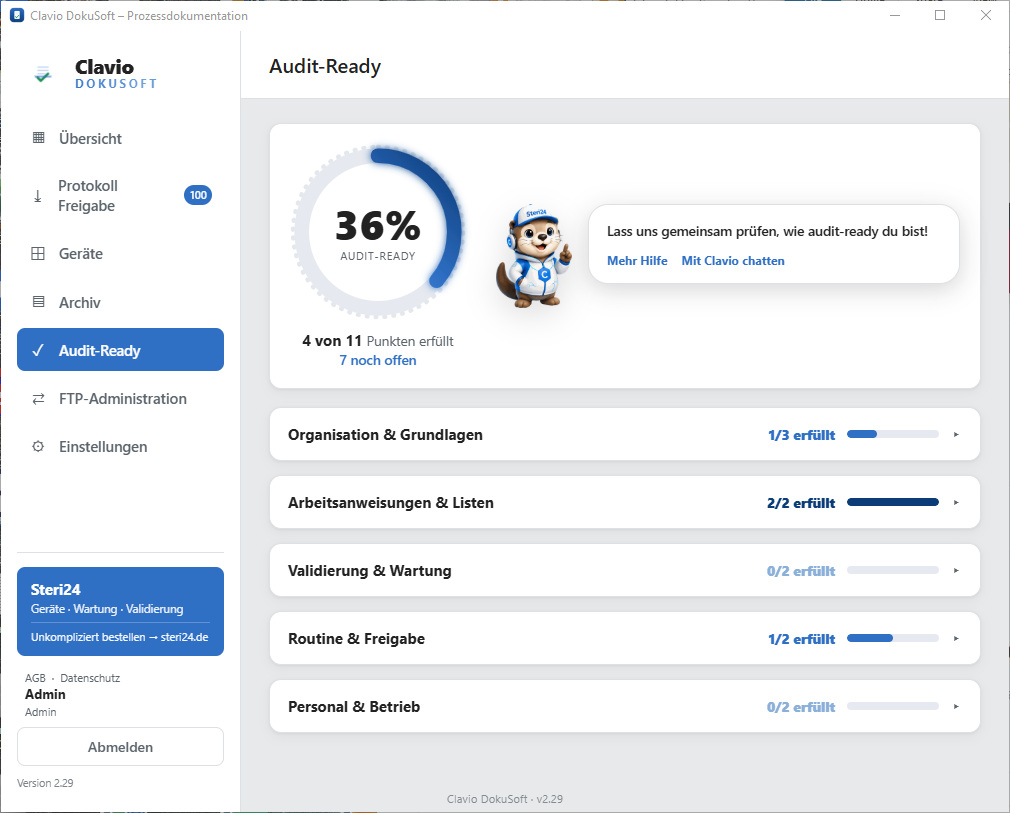
.png "Preview: Clavio DokuSoft")



Frequently Asked Questions
RKI Legal Requirements – key questions answered clearly.