RKI wettelijke vereisten
 RKI 1: Wettelijke vereisten & overzicht
RKI 1: Wettelijke vereisten & overzicht  RKI 2: De correcte voorreiniging
RKI 2: De correcte voorreiniging  RKI 3: Reiniging & desinfectie
RKI 3: Reiniging & desinfectie 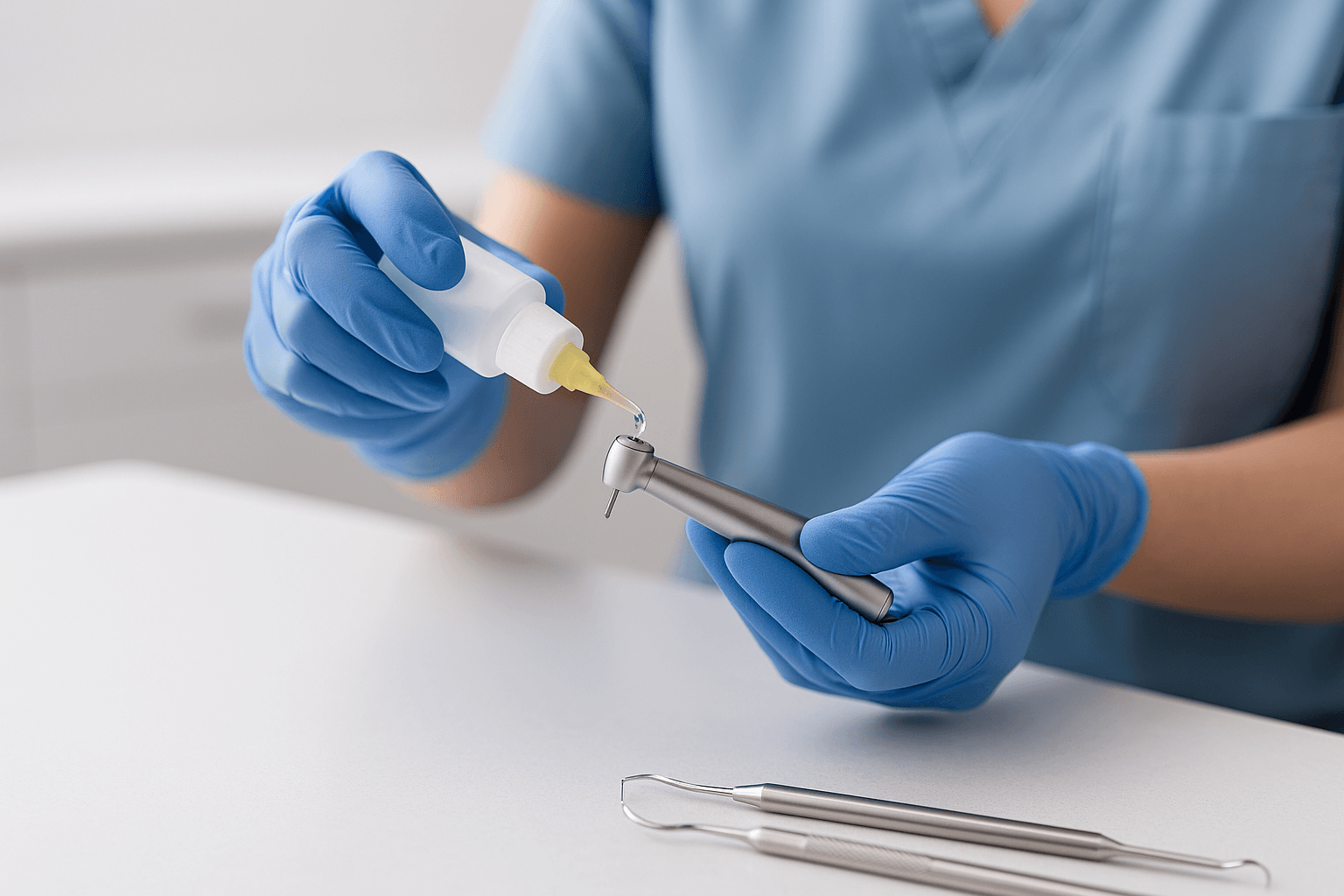 RKI 4: Onderhoud van de instrumenten
RKI 4: Onderhoud van de instrumenten 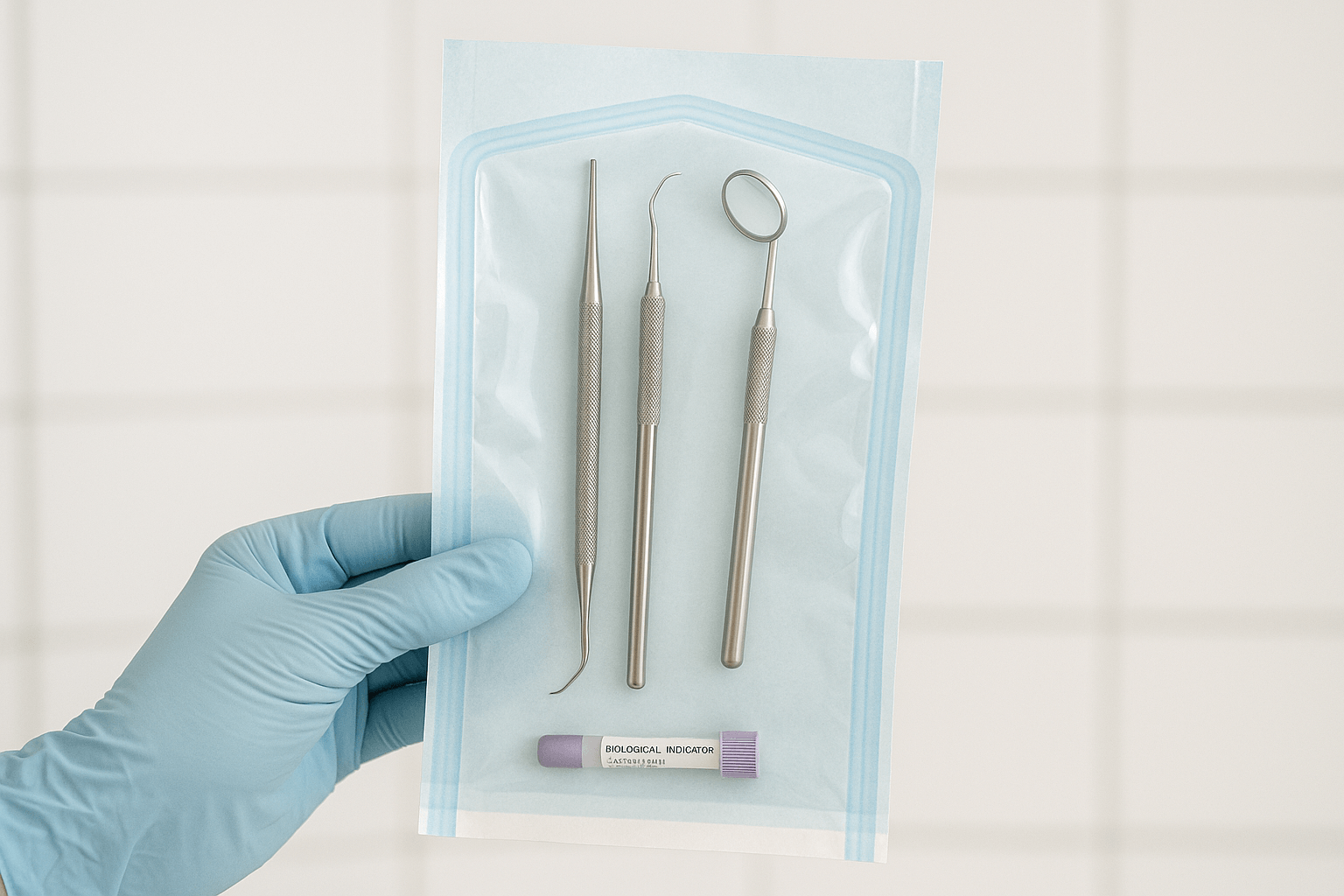 RKI 5: Verpakking van de instrumenten
RKI 5: Verpakking van de instrumenten  RKI 6: Sterilisatie in de autoclaaf
RKI 6: Sterilisatie in de autoclaaf  RKI 7: Vrijgave & documentatie
RKI 7: Vrijgave & documentatie  RKI 8: Opslag van steriele instrumenten
RKI 8: Opslag van steriele instrumenten 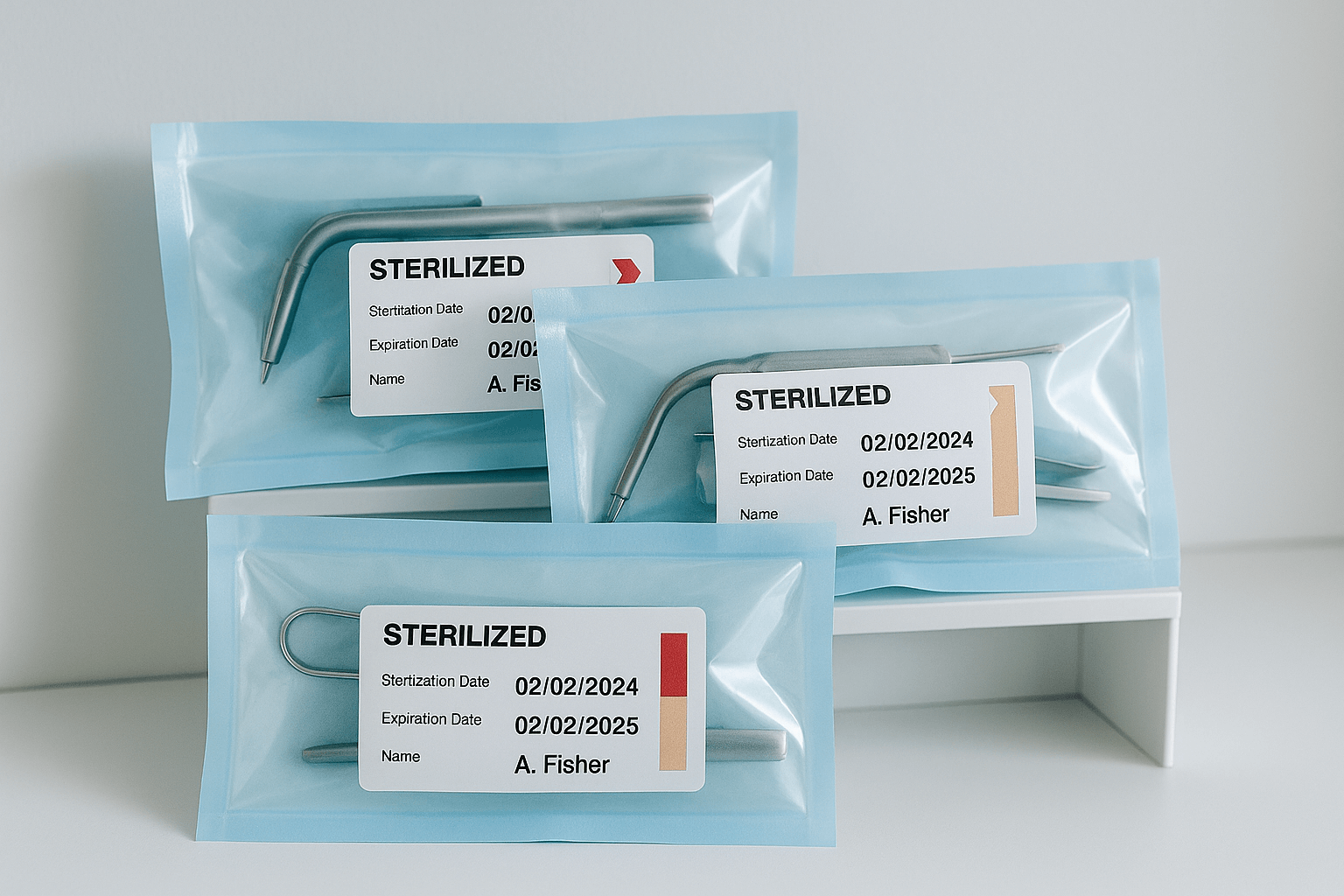 RKI 9: Markering na sterilisatie
RKI 9: Markering na sterilisatie 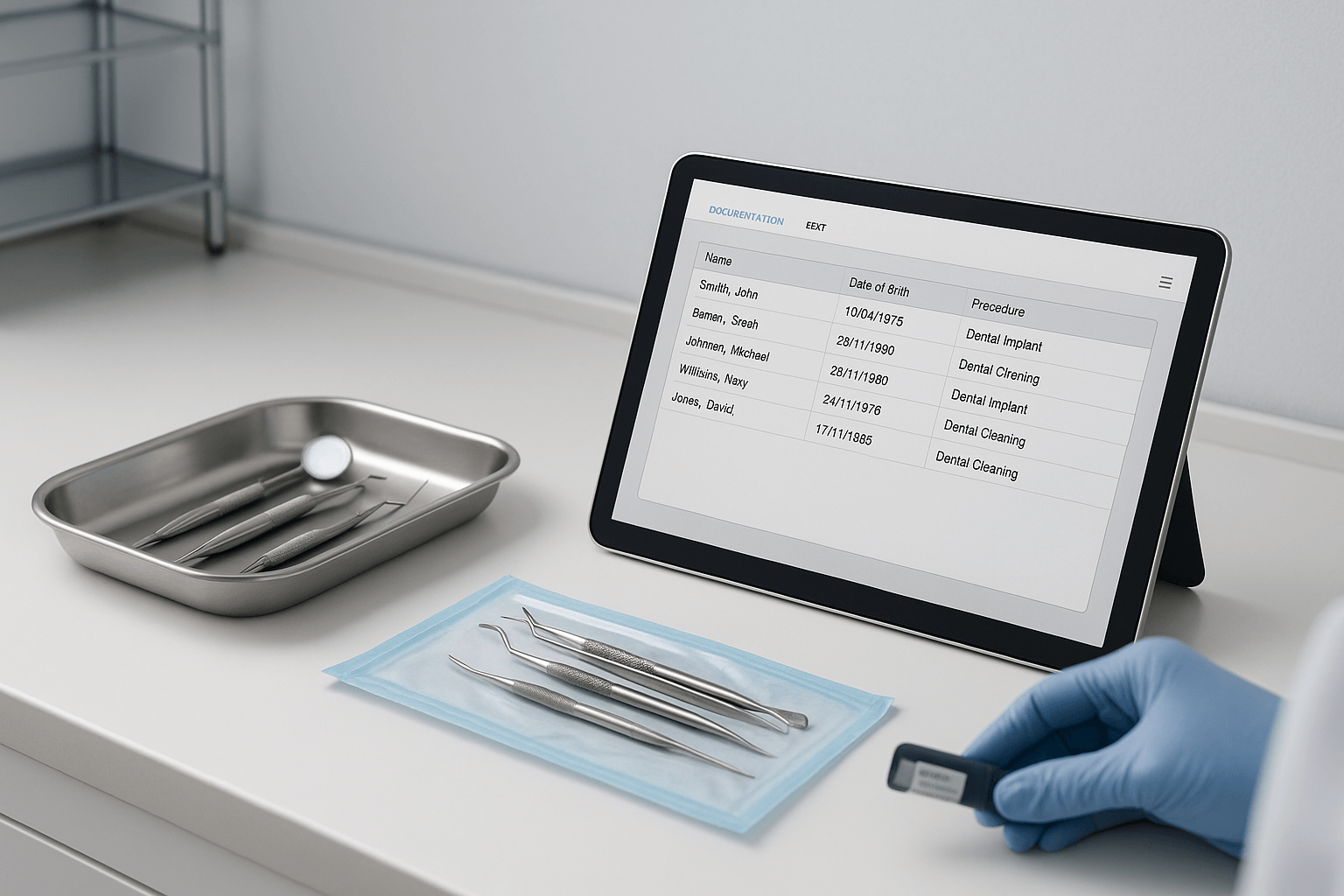 RKI 10: Documentatie na gebruik bij de patiënt
RKI 10: Documentatie na gebruik bij de patiënt  RKI 11: Hygiëneruimte gemäß KRINKO
RKI 11: Hygiëneruimte gemäß KRINKO  RKI 12: Eerste validatie autoclaaf klasse B
RKI 12: Eerste validatie autoclaaf klasse B
Wettelijke vereisten & risicoclassificatie
Alle belangrijke documenten in één oogopslag om te downloaden en af te drukken:
KRINKO-aanbeveling verwerking medische hulpmiddelen
Beladingspatroon autoclaaf
Chargevrijgave handgeschreven
Voorbeeld zeeflijst
Standaard werkinstructies verwerking kritische B-instrumenten
Sjabloon risicobeoordeling van de instrumenten
Tandartsenkamer werkinstructies voor de verwerking van medische hulpmiddelen
De praktijkexploitant draagt de verantwoordelijkheid dat de wettelijke vereisten worden nageleefd.
1) Wet inzake infectiebescherming ter infectiepreventie (IfSG)
2) De wet op de medische hulpmiddelen bepaalt hoe met medische hulpmiddelen moet worden omgegaan (MDR)
3) De verordening voor exploitanten van medische hulpmiddelen verwijst naar KRINKO (MPBetreibV)
4) Aanbevelingen van de KRINKO bij het RKI beschrijven de precieze verwerkingsprocedure stap voor stap
In § 8 van de MPBetreibV wordt het volgende vereist:
1) De aanwijzingen van de fabrikant moeten worden nageleefd
2) De procedure voor de instrumentenverwerking moet gevalideerd zijn
3) Bewijsplicht, waaraan door middel van documentatie kan worden voldaan
4) Aanbevelingen van het RKI moeten in acht worden genomen
5) De gebruiker en validator moeten geschoold zijn volgens § 5
a. Ofwel door een opleiding in het beroep
b. Scholing medisch toestel
Verschaf u eerst een overzicht
Stel een lijst op van alle producten die verwerkt moeten worden
Stel een lijst op van alle instrumenten die u meermaals wilt gebruiken.
Bijvoorbeeld als volgt:
| Naam | Aantal (fictief) |
|---|---|
| ECG-elektroden | 12 |
| Oortrechter | 5 |
| Oorspoelspuit | 2 |
| Speculum | 3 |
| Flexibele endoscopen (bijv. gastroscoop) | 2 |
| Schaar | 5 |
| Chirurgisch pincet | 5 |
| Anatomisch pincet | 5 |
| Wondhaak | 5 |
| Pean-klem | 5 |
| Scherpe lepel | 5 |
| Naaldvoerder | 5 |
| MIC-trocart | 4 |
| ERCP-katheter | 2 |
Toewijzing van de correcte risicoklasse aan elk instrument
De vakkundige verwerking van medische hulpmiddelen is doorslaggevend voor de patiëntveiligheid en vereist een nauwkeurige risicobeoordeling en indeling van de instrumenten. Volgens de KRINKO-aanbeveling van het Robert Koch-Instituut (RKI) worden medische hulpmiddelen op basis van hun contact met de patiënt onderverdeeld in drie risicocategorieën:
1) Niet-kritische medische hulpmiddelen
2) Semikritische medische hulpmiddelen
3) Kritische medische hulpmiddelen
Binnen de categorieën "semikritisch" en "kritisch" wordt bovendien onderscheid gemaakt tussen de klassen A, B en C, afhankelijk van de specifieke eisen aan de verwerking.
| Definitie | Risicoklasse | Specifieke klasse |
|---|---|---|
| Medische hulpmiddelen die enkel met intacte huid in aanraking komen | Niet-kritische medische hulpmiddelen | Niet-kritisch |
| Medische hulpmiddelen die met slijmvlies of pathologisch veranderde huid in aanraking komen | Semikritische medische hulpmiddelen (A of B) |
Semikritisch A: Medische hulpmiddelen zonder voor de stoom moeilijk te bereiken plaatsen |
|
Semikritisch B: Medische hulpmiddelen met holle ruimten of andere voor de stoom moeilijk te bereiken plaatsen |
||
|
Medische hulpmiddelen die volgens hun bestemming de huid of het slijmvlies doordringen en daarbij in contact komen met bloed, respectievelijk bij inwendige weefsels of organen worden toegepast. Medische hulpmiddelen voor de toediening van bloed, bloedproducten of andere steriele geneesmiddelen/steriele medische hulpmiddelen Medische hulpmiddelen die in het lichaam in contact komen met urine. |
Kritische medische hulpmiddelen (A, B of C) | Kritisch A: Medische hulpmiddelen zonder voor de stoom moeilijk te bereiken plaatsen |
| Kritisch B: Medische hulpmiddelen met holle ruimten of andere voor de stoom moeilijk te bereiken plaatsen |
||
| Kritisch C: Volgens de aanwijzingen van de fabrikant met bijzonder hoge eisen aan de verwerking (bijvoorbeeld kritisch B medische hulpmiddelen die niet met stoom gesteriliseerd kunnen worden) |
Hierna een voorbeeld van hoe een opsomming en classificatie van medische hulpmiddelen kan verlopen:
| Naam | Aantal (fictief) | Risico-indeling volgens RKI |
|---|---|---|
| ECG-elektroden | 12 | Niet-kritisch |
| Oortrechter | 5 | Semikritisch A |
| Oorspoelspuit | 2 | Semikritisch A |
| Speculum | 3 | Semikritisch A |
| Flexibele endoscopen (bijv. gastroscoop) | 2 | Semikritisch B |
| Schaar | 5 | Kritisch A |
| Chirurgisch pincet | 5 | Kritisch A |
| Anatomisch pincet | 5 | Kritisch A |
| Wondhaak | 5 | Kritisch A |
| Pean-klem | 5 | Kritisch A |
| Scherpe lepel | 5 | Kritisch A |
| Naaldvoerder | 5 | Kritisch A |
| MIC-trocart | 4 | Kritisch B |
| ERCP-katheter | 2 | Kritisch C |
Soms is de indeling niet eenduidig. Zo zou een schaar ook als kritisch B ingedeeld kunnen worden, aangezien het scharnier moeilijk te reinigen is. Eveneens zou een gastroscoop als kritisch B ingedeeld kunnen worden, aangezien deze vaak met bloed in contact komt (bijv. in het kader van een biopsie). Door sommige artsenverenigingen worden op het internet brochures ter beschikking gesteld die zeer gedetailleerd en deels geïllustreerd de actuele eisen begrijpelijk weergeven.
Werkinstructies voor de verwerking van medische hulpmiddelen - Tandartsenkamer Berlijn
De noodzakelijke verwerkingsstappen voor elke risicoklasse
| Risicoklasse | Verwerking |
|---|---|
| Niet-kritisch | Voorreiniging: Nee Reiniging met desinfectie: Ja, manueel met de hand Verpakken: Nee Sterilisatie in de autoclaaf: Nee Markering: Nee |
| Semikritisch A | Voorreiniging: Nee, optioneel. Reiniging met desinfectie: Ja. Manueel met de hand, of met een ultrasoonreiniger of door middel van een thermodesinfector. Aanbeveling: ultrasoonreiniger Verpakken: Nee Sterilisatie in de autoclaaf: Enkel optioneel. Markering: Nee |
| Semikritisch B | Voorreiniging: Ja, de voorreiniging moet onmiddellijk na het gebruik worden uitgevoerd. Bijvoorbeeld inleggen in een Reiniging met thermische desinfectie: Ja, manueel wanneer machinaal niet mogelijk is. Anders machinaal met ultrasoonreiniger of thermodesinfector. Verpakken: Nee Sterilisatie in de autoclaaf: Verplicht voor instrumenten die in een steriele lichaamsholte worden gebruikt. Bijv. de urineblaas. Markering: Nee |
| Kritisch A | Voorreiniging: Nee, optioneel. Reiniging met desinfectie: Ja, ofwel in de ultrasoonreiniger ofwel de thermodesinfector. Verpakken: Ja Sterilisatie in de autoclaaf: Ja, verpakt. Markering: Ja |
| Kritisch B | Voorreiniging: Ja, onmiddellijk na het gebruik. Reiniging met desinfectie: Ja, in de thermodesinfector. Verpakken: Ja Sterilisatie in de autoclaaf: Ja, verpakt. Markering: Ja |
| Kritisch C | Voorreiniging: Volgens de aanwijzingen van de fabrikant van het instrument. Reiniging met desinfectie: Volgens de aanwijzingen van de fabrikant van het instrument. Verpakken: Ja Sterilisatie in de autoclaaf: Volgens de aanwijzingen van de fabrikant van het instrument. Markering: Ja |
Nu kan een volledige lijst van alle instrumenten worden opgesteld met een overzicht van welke toestellen gebruikt moeten worden:
Volledig overzicht van de instrumenten en toestellen
| Naam | Aantal (fictief) | Risico-indeling | Voorreiniging | Reiniging + desinfectie | Markering | Sterilisatie |
|---|---|---|---|---|---|---|
| ECG-elektroden | 12 | Niet-kritisch | Nee | Ja | Nee | Nee |
| Oortrechter | 5 | Semikritisch A | Optioneel | Ja | Nee | Optioneel |
| Oorspoelspuit | 2 | Semikritisch A | Optioneel | Ja | Nee | Optioneel |
| Speculum | 3 | Semikritisch A | Optioneel | Ja | Nee | Optioneel |
| Flexibele endoscopen (bijv. gastroscoop) | 2 | Semikritisch B | Ja | Ja | Nee | Bij gebruik in steriele lichaamsholten |
| Schaar | 5 | Kritisch A | Optioneel | Ja | Nee | Ja |
| Chirurgisch pincet | 5 | Kritisch A | Optioneel | Ja | Nee | Ja |
| Anatomisch pincet | 5 | Kritisch A | Optioneel | Ja | Nee | Ja |
| Wondhaak | 5 | Kritisch A | Optioneel | Ja | Nee | Ja |
| Pean-klem | 5 | Kritisch A | Optioneel | Ja | Nee | Ja |
| Scherpe lepel | 5 | Kritisch A | Optioneel | Ja | Nee | Ja |
| Naaldvoerder | 5 | Kritisch A | Optioneel | Ja | Nee | Ja |
| MIC-trocart | 4 | Kritisch B | Ja | Ja | Optioneel | Ja |
| ERCP-katheter | 2 | Kritisch C | Ja | Ja | Ja | Ja |
Hoe audit-ready is uw praktijk?
Controleer met de gratis audit-check van ClavioSoft of uw instrumentenverwerking aan alle RKI-vereisten voldoet — categorie per categorie, met duidelijke voortgang en concrete openstaande punten.
.png "Preview: Clavio DokuSoft")



Häufige Fragen zur Instrumentenaufbereitung nach RKI
Die wichtigsten Fragen und fachlich fundierten Antworten zur Instrumentenaufbereitung nach RKI – auf Basis von RKI/KRINKO und den einschlägigen Normen.