RKI Gesetzliche Anforderungen
 RKI 1: Gesetzliche Anforderungen & Überblick
RKI 1: Gesetzliche Anforderungen & Überblick  RKI 2: Die korrekte Vorreinigung
RKI 2: Die korrekte Vorreinigung  RKI 3: Reinigung & Desinfektion
RKI 3: Reinigung & Desinfektion  RKI 4: Pflege der Instrumente
RKI 4: Pflege der Instrumente  RKI 5: Verpackung der Instrumente
RKI 5: Verpackung der Instrumente  RKI 6: Sterilisation im Autoklav
RKI 6: Sterilisation im Autoklav  RKI 7: Freigabe & Dokumentation
RKI 7: Freigabe & Dokumentation  RKI 8: Lagerung steriler Instrumente
RKI 8: Lagerung steriler Instrumente 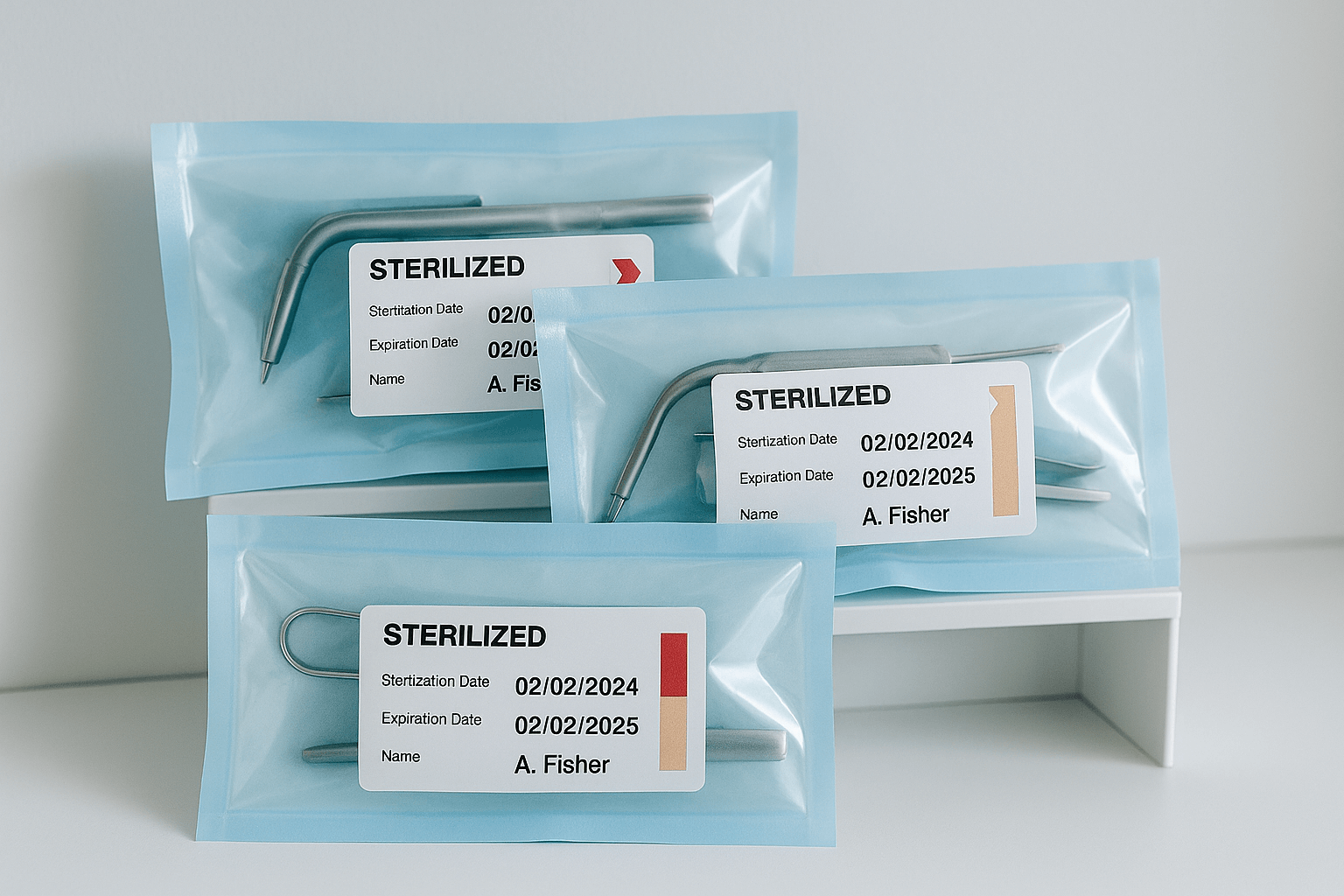 RKI 9: Kennzeichnung nach Sterilisation
RKI 9: Kennzeichnung nach Sterilisation  RKI 10: Dokumentation nach Patienteneinsatz
RKI 10: Dokumentation nach Patienteneinsatz  RKI 11: Hygieneraum gemäß KRINKO
RKI 11: Hygieneraum gemäß KRINKO  RKI 12: Erstvalidierung Autoklav Klasse B
RKI 12: Erstvalidierung Autoklav Klasse B
RKI-Auflagen erfüllen – kostenlos
Machen Sie mit der kostenlosen ClavioSoft in wenigen Minuten einen Audit-Check Ihrer Praxis – fehlende Dokumentation erstellt die Software gleich mit. Das nimmt Ihnen bis zu 90 % der Arbeit ab.

Instrumentenaufbereitung nach RKI: gesetzliche Anforderungen & Risikoklassen
In Deutschland ist die Aufbereitung wiederverwendbarer Medizinprodukte gesetzlich verpflichtend und klar geregelt. Wer Instrumente mehrfach benutzt, muss sie mit einem validierten Verfahren aufbereiten, die Empfehlung der KRINKO beim Robert Koch-Institut (RKI) beachten und jeden Schritt nachweisbar dokumentieren. Die Verantwortung dafür trägt immer der Praxisbetreiber (§ 8 MPBetreibV).
Diese Seite ist der Einstieg in unsere 12-teilige Schritt-für-Schritt-Anleitung zur RKI-konformen Instrumentenaufbereitung. Sie erklärt – auch ohne Vorwissen –, welche Gesetze gelten, wie Sie jedes Ihrer Instrumente in die richtige Risikoklasse einordnen und welches Aufbereitungsverfahren daraus zwingend folgt. Am Ende wissen Sie genau, was in Ihrer Praxis zu tun ist.
1Was verlangt der Gesetzgeber?
Vier Regelwerke greifen ineinander. Sie müssen die Gesetze nicht auswendig kennen – wichtig ist nur zu verstehen, worauf sie hinauslaufen:
| Regelwerk | Was es regelt | Was das für Sie bedeutet |
|---|---|---|
| IfSG Infektionsschutzgesetz | Grundlegender Schutz vor Infektionen in Einrichtungen des Gesundheitswesens. | Sie müssen Übertragungen von Keimen aktiv verhindern – saubere Instrumente sind Pflicht. |
| MDR & MPDG EU-Medizinprodukteverordnung 2017/745 + Durchführungsgesetz | Lösen seit 2021 das alte Medizinproduktegesetz (MPG) ab und regeln den sicheren Umgang mit Medizinprodukten. | Wiederverwendbare Produkte dürfen nur so aufbereitet werden, dass ihre Sicherheit und Funktion erhalten bleiben. |
| MPBetreibV Medizinprodukte-Betreiberverordnung, § 8 | Konkretisiert, wie aufbereitet werden muss – und verweist dafür ausdrücklich auf die KRINKO/RKI-Empfehlung. | Das zentrale Pflichtenheft für Ihre Praxis (siehe unten). |
| KRINKO/RKI Empfehlung „Anforderungen an die Hygiene bei der Aufbereitung von Medizinprodukten“ | Beschreibt das eigentliche Aufbereitungsverfahren Schritt für Schritt. | Ihre konkrete Arbeitsanleitung – genau sie setzen wir in den 12 Kapiteln um. |
Die Kernpflichten aus § 8 MPBetreibV
Der entscheidende Paragraf ist § 8 der MPBetreibV. Er fordert von Ihnen als Betreiber konkret:
1) Herstellerangaben beachten – jedes Instrument so aufbereiten, wie es der Hersteller freigibt (vgl. DIN EN ISO 17664).
2) Validiertes Verfahren – Reinigung, Desinfektion und Sterilisation müssen nachweislich wirksam und reproduzierbar sein.
3) Nachweispflicht – der Erfolg jeder Aufbereitung ist zu dokumentieren (Chargendokumentation). Diese Chargen- und Freigabedokumentation erledigen Sie mit der kostenlosen ClavioSoft in wenigen Klicks – inkl. revisionssicherer Ablage.
4) KRINKO/RKI-Empfehlung befolgen – sie ist der anerkannte Stand von Wissenschaft und Technik.
5) Geschultes Personal – wer aufbereitet, braucht die nötige Sachkenntnis (Ausbildung oder Schulung; vgl. KRINKO-Anlage 6).
2So gehen Sie in der Praxis vor – in 4 Schritten
Bevor Sie ein einziges Instrument aufbereiten, legen Sie schriftlich fest, ob, womit und unter welchen Bedingungen jedes Produkt aufbereitet wird. Das verlangt die KRINKO ausdrücklich – diese Festlegung ist zugleich ein zentraler Baustein Ihres Hygieneplans und einfacher umzusetzen, als es klingt:
Quelle: KRINKO/BfArM-Empfehlung 2012, Abschn. 1.2.1 „Risikobewertung und Einstufung“.Schritt 1: Liste aller wiederverwendbaren Instrumente
Erstellen Sie zunächst eine vollständige Liste aller Produkte, die Sie mehrfach verwenden möchten – ganz ohne Wertung, einfach alles auflisten. Zum Beispiel:
| Name | Anzahl (Beispiel) |
|---|---|
| EKG-Elektroden | 12 |
| Ohrtrichter | 5 |
| Ohrspülspritze | 2 |
| Spekulum | 3 |
| Flexible Endoskope (z. B. Gastroskop) | 2 |
| Schere | 5 |
| Chirurgische Pinzette | 5 |
| Anatomische Pinzette | 5 |
| Wundhaken | 5 |
| Pean-Klemme | 5 |
| Scharfer Löffel | 5 |
| Nadelhalter | 5 |
| MIC-Trokar | 4 |
| ERCP-Katheter | 2 |
Schritt 2: Jedem Instrument die Risikoklasse zuordnen
Die Risikoklasse richtet sich danach, womit das Instrument am Patienten in Kontakt kommt. Die KRINKO unterscheidet drei Hauptklassen:
| Kontakt mit dem Patienten | Risikoklasse | Feinunterteilung |
|---|---|---|
| Nur Kontakt mit intakter Haut | Unkritisch | – |
| Kontakt mit Schleimhaut oder krankhaft veränderter Haut | Semikritisch (A oder B) | A: ohne für Dampf schwer erreichbare Stellen |
| B: mit Hohlräumen / schwer erreichbaren Stellen | ||
| Durchdringt Haut/Schleimhaut und kommt mit Blut, inneren Geweben oder Organen in Kontakt (auch: Anwendung von Blut/sterilen Arzneimitteln; Kontakt mit Urin im Körper) | Kritisch (A, B oder C) | A: ohne für Dampf schwer erreichbare Stellen |
| B: mit Hohlräumen / schwer erreichbaren Stellen | ||
| C: besonders hohe Anforderungen lt. Hersteller (z. B. nicht dampfsterilisierbar) |
So sieht die Einstufung am Beispiel aus:
| Name | Anzahl (Beispiel) | Risikoeinstufung nach RKI |
|---|---|---|
| EKG-Elektroden | 12 | Unkritisch |
| Ohrtrichter | 5 | Semikritisch A |
| Ohrspülspritze | 2 | Semikritisch A |
| Spekulum | 3 | Semikritisch A |
| Flexible Endoskope (z. B. Gastroskop) | 2 | Semikritisch B |
| Schere | 5 | Kritisch A |
| Chirurgische Pinzette | 5 | Kritisch A |
| Anatomische Pinzette | 5 | Kritisch A |
| Wundhaken | 5 | Kritisch A |
| Pean-Klemme | 5 | Kritisch A |
| Scharfer Löffel | 5 | Kritisch A |
| Nadelhalter | 5 | Kritisch A |
| MIC-Trokar | 4 | Kritisch B |
| ERCP-Katheter | 2 | Kritisch C |
Passenden Klasse-B-Autoklaven finden
Schritt 3: Pro Risikoklasse das richtige Verfahren festlegen
Aus der Risikoklasse ergibt sich, welche Aufbereitungsschritte Pflicht sind – von der Vorreinigung bis zur Sterilisation. Diese Tabelle übersetzt jede Klasse in konkrete Handlungen:
| Risikoklasse | Aufbereitung |
|---|---|
| Unkritisch | Vorreinigung: nein · Reinigung und Desinfektion: ja (manuell oder maschinell) mit nachweislich wirksamem Desinfektionsmittel bzw. -verfahren; maschinell ist wegen der besseren Reproduzierbarkeit zu bevorzugen · Verpacken: nein · Sterilisation: nein · Kennzeichnung: nein |
| Semikritisch A | Vorreinigung: optional · Reinigung und anschließend wirksame Desinfektion mit nachgewiesenem Wirkungsbereich (bakterizid einschließlich Mykobakterien, fungizid, viruzid) – manuell oder maschinell. Ein Ultraschallreiniger unterstützt nur die Reinigung und ersetzt die Desinfektion nicht; der Thermodesinfektor (RDG) leistet Reinigung und Desinfektion in einem Schritt. · Verpacken: nein · Sterilisation: nur optional · Kennzeichnung: nein |
| Semikritisch B | Vorreinigung: ja, unmittelbar nach Anwendung (z. B. einlegen) · Reinigung und Desinfektion bevorzugt maschinell/thermisch im Thermodesinfektor (RDG); Ultraschall ist allenfalls ein vorgeschalteter Reinigungsschritt und kein Desinfektionsverfahren. Manuell nur, wenn maschinell nicht möglich · Verpacken: nein · Sterilisation: verpflichtend bei Einsatz in steriler Körperhöhle · Kennzeichnung: nein |
| Kritisch A | Vorreinigung: optional · Reinigung und Desinfektion: bevorzugt maschinell im Thermodesinfektor (RDG); ein Ultraschallreiniger dient nur der unterstützenden Reinigung (z. B. Vorreinigung) und ersetzt die Desinfektion nicht · Verpacken: ja · Sterilisation: ja, verpackt · Kennzeichnung: ja |
| Kritisch B | Vorreinigung: ja, unmittelbar nach Anwendung · Reinigung+Desinfektion: ja, im Thermodesinfektor · Verpacken: ja · Sterilisation: ja, verpackt (Klasse B) · Kennzeichnung: ja |
| Kritisch C | Vorreinigung / Reinigung+Desinfektion / Sterilisation: jeweils gemäß Herstellerangaben des Instruments · Verpacken: ja · Kennzeichnung: ja |
Schritt 4: Alles in einer Gesamtübersicht zusammenführen
Zum Schluss verbinden Sie beide Tabellen zu einer Übersicht. Sie zeigt für jedes Instrument auf einen Blick, welche Geräte und Schritte nötig sind – und ist zugleich Ihre dokumentierte Festlegung im Sinne der KRINKO:
| Name | Anzahl | Risikoeinstufung | Vorreinigung | Reinigung + Desinfektion | Kennzeichnung | Sterilisation |
|---|---|---|---|---|---|---|
| EKG-Elektroden | 12 | Unkritisch | Nein | Ja | Nein | Nein |
| Ohrtrichter | 5 | Semikritisch A | Optional | Ja | Nein | Optional |
| Ohrspülspritze | 2 | Semikritisch A | Optional | Ja | Nein | Optional |
| Spekulum | 3 | Semikritisch A | Optional | Ja | Nein | Optional |
| Flexible Endoskope (z. B. Gastroskop) | 2 | Semikritisch B | Ja | Ja | Nein | Bei Einsatz in steriler Körperhöhle |
| Schere | 5 | Kritisch A | Optional | Ja | Nein | Ja |
| Chirurgische Pinzette | 5 | Kritisch A | Optional | Ja | Nein | Ja |
| Anatomische Pinzette | 5 | Kritisch A | Optional | Ja | Nein | Ja |
| Wundhaken | 5 | Kritisch A | Optional | Ja | Nein | Ja |
| Pean-Klemme | 5 | Kritisch A | Optional | Ja | Nein | Ja |
| Scharfer Löffel | 5 | Kritisch A | Optional | Ja | Nein | Ja |
| Nadelhalter | 5 | Kritisch A | Optional | Ja | Nein | Ja |
| MIC-Trokar | 4 | Kritisch B | Ja | Ja | Optional | Ja |
| ERCP-Katheter | 2 | Kritisch C | Ja | Ja | Ja | Ja |
3Vorlagen & Dokumente zum Download
Diese Vorlagen und Original-Quellen helfen Ihnen, die oben beschriebenen Schritte direkt umzusetzen – zum Ausdrucken und Anpassen an Ihre Praxis:
Wichtige Zusatzpunkte gemäß RKI & Normen
Diese Punkte werden in der Praxis häufig übersehen, sind aber für die korrekte und rechtssichere Umsetzung wichtig:
- Erweiterung der genannten Normen je Verfahrensschritt: Ergaenzen Sie neben DIN EN 13060 und DIN EN ISO 17664 die fuer die einzelnen Schritte einschlaegigen Normen: DIN EN ISO 15883 (Reinigungs- und Desinfektionsgeraete/Thermodesinfektion), DIN EN ISO 17665 (Validierung der Dampfsterilisation) sowie DIN EN ISO 11607-1/-2 und DIN 58953 (Sterilbarrieresysteme/Verpackung). Diese Normen sind im Normenanhang der KRINKO/BfArM-Empfehlung 2012 als mitgeltend aufgefuehrt.Quelle: KRINKO/BfArM 2012, Normenanhang (nennt DIN EN ISO 15883, 17665, 11607, DIN 58953)
- Vorbehandlung/Sammeln am Gebrauchsort: Ergaenzen Sie die sachgerechte Vorbehandlung am Gebrauchsort als ersten Schritt: grobe Verschmutzungen unmittelbar nach Anwendung entfernen, ein Antrocknen von Blut/Gewebe vermeiden (z. B. Abwischen, Spuelen von Arbeitskanaelen, Festlegung von Entsorgungszeiten) und fixierende Verfahren vor der Reinigung vermeiden. Die KRINKO zaehlt Vorbehandlung, Sammlung und Vorreinigung ausdruecklich zum ersten Arbeitsschritt der Aufbereitung.Quelle: KRINKO/BfArM 2012, Abschn. 2.2.1 'Vorbereitung der Aufbereitung (Vorbehandlung, Sammlung, Vorreinigung ...)'
- Freigabe zur Anwendung als eigener, dokumentierter Schritt: Ergaenzen Sie die dokumentierte Freigabe zur Anwendung als Abschluss der Aufbereitung: Abgleich der ermittelten Prozessparameter mit den Validierungsberichten, Durchfuehrung und Dokumentation der Routinepruefungen, Pruefung der Verpackung auf Unversehrtheit und Trockenheit sowie der Kennzeichnung; die zur Freigabe berechtigten Personen sind schriftlich zu benennen. Erst danach darf das Sterilgut verwendet werden. Die dazugehoerige Freigabe-Dokumentation fuehren Sie mit der kostenlosen ClavioSoft nachvollziehbar und papierlos.Quelle: KRINKO/BfArM 2012, Abschn. 2.2.7 'Freigabe zur Anwendung'; MPBetreibV § 8
- Validierung der Verfahren (Erst- und Re-Validierung) konkretisieren: Der Artikel nennt 'validiertes Verfahren', erklaert aber dessen Umsetzung nicht: Reinigungs-/Desinfektions- und Sterilisationsprozesse sind vor der ersten Nutzung zu validieren und regelmaessig erneut zu pruefen; Aenderungen an Beladung, Programm oder Verpackung erfordern eine erneute Bewertung. Konkrete Pruefintervalle ergeben sich aus den Hersteller- bzw. Normvorgaben (keine pauschale Frist nennen).Quelle: KRINKO/BfArM 2012, Abschn. 1.3 'Validierung'; Normenanhang (DIN EN ISO 17665, DIN EN ISO 15883)
- Arbeitstaegliche Routinepruefungen und Indikatoren im Sterilisationsbetrieb: Ergaenzen Sie die laufenden Routinepruefungen des Klasse-B-Autoklaven: Vakuum-/Dichtheitstest und Pruefung der Dampfdurchdringung mit geeignetem Pruefkoerper (Hohlkoerper) sowie Chargenkontrolle mit Behandlungsindikatoren. Diese Pruefungen sind Voraussetzung der Chargenfreigabe.Quelle: DIN EN 13060 (Pruefungen Klasse-B-Sterilisator, Anhang B); KRINKO/BfArM 2012, Abschn. 2.2.7
- Wasserqualitaet fuer Dampfsterilisation und maschinelle Aufbereitung: Ergaenzen Sie die Anforderung an die Wasserqualitaet: Fuer den Dampf-Kleinsterilisator ist Speisewasser entsprechend DIN EN 13060 (Anhang C) erforderlich; auch fuer die Schlussspuelung im Thermodesinfektor wird aufbereitetes Wasser benoetigt, um Flecken, Korrosion und Geraeteschaeden zu vermeiden.Quelle: KRINKO/BfArM 2012, Anlage 4 'Speisewasserqualitaet: DIN EN 13060, Anhang C'
- Bauliche/zonale Trennung rein/unrein und Personalschutz: Ergaenzen Sie die KRINKO-Forderung nach getrennten Arbeitsbereichen (unrein: Annahme/Reinigung – rein: Verpackung/Freigabe – Lagerung) sowie persoenliche Schutzausruestung im unreinen Bereich (geeignete Handschuhe, Schutzbrille, Schutzkleidung), um Kreuzkontamination und Personalgefaehrdung zu vermeiden.Quelle: KRINKO/BfArM 2012, Anlage 5 (Zonen-/Bereichstrennung unrein-rein-Lagerung) sowie Abschn. 2.2.1 (Arbeitsschutz, TRBA 250)
- Kontrolle des Reinigungserfolgs (Sauberkeit/Restproteine): Ergaenzen Sie die Kontrolle des Reinigungserfolgs: Sicht-/Lupenkontrolle auf Sauberkeit und Unversehrtheit; beim nicht-maschinellen (manuellen) Verfahren ist der Reinigungserfolg gesondert abzusichern (z. B. stichprobenartige Proteinbestimmung; KRINKO nennt einen Warnwert von 100 µg Protein pro Instrument).Quelle: KRINKO/BfArM 2012, Abschn. 2.2.2 (Pruefung auf Sauberkeit; Warnwert 100 µg Protein)
- Nach Risikoklasse differenzierte Sachkenntnis: Praezisieren Sie die Personalanforderungen: Fuer 'kritisch B' fordert die KRINKO zusaetzlich den Nachweis einer anerkannten Ausbildung der mit der Aufbereitung betrauten Person; fuer 'kritisch C' kommt eine Zertifizierung des Qualitaetsmanagementsystems (DIN EN ISO 13485 durch eine anerkannte Stelle) hinzu.Quelle: KRINKO/BfArM 2012, Tab. 1 (Fussnoten zu kritisch B/C) und Anlage 6 'Sachkenntnis des Personals'; DIN EN ISO 13485
Benötigte Geräte, Verbrauchsmaterialien & Zubehör
Checkliste für diesen Arbeitsschritt – das sollten Sie bereithalten (Einkaufsliste):
Geräte
- Klasse-B-Dampf-Kleinsterilisator (Autoklav) nach DIN EN 13060 fuer die verpackte Sterilisation kritischer Instrumente Hier im Shop bestellen
- Thermodesinfektor (Reinigungs- und Desinfektionsgeraet, RDG) nach DIN EN ISO 15883 fuer die bevorzugte maschinelle Reinigung und Desinfektion Hier im Shop bestellen
- Ultraschallreiniger als unterstuetzendes Reinigungs-/Vorreinigungsgeraet (ersetzt keine Desinfektion) Hier im Shop bestellen
- Wasseraufbereitung/Entsalzung fuer demineralisiertes Speise- und Spuelwasser Hier im Shop bestellen
- Siegelgeraet fuer Klarsicht-Sterilisierverpackungen (validierbar, nach DIN EN ISO 11607-2) Hier im Shop bestellen
- Digitale Dokumentationssoftware bzw. Drucker/Datenlogger zur Chargen- und Freigabedokumentation Hier im Shop bestellen Digital mit ClavioSoft erstellen
Verbrauchsmaterialien
- Reinigungs- und Desinfektionsmittel mit nachgewiesenem Wirkungsbereich (bakterizid inkl. Mykobakterien, fungizid, viruzid) Hier im Shop bestellen
- Klarsicht-Sterilisierverpackung/Sterilbarrieresystem und Vlies/Krepp nach DIN EN ISO 11607-1 / DIN 58953 Hier im Shop bestellen
- Behandlungs-/Chemoindikatoren und Indikatorband zur Chargenkontrolle Hier im Shop bestellen
- Pruefkoerper/Testsystem fuer Vakuum- und Dampfdurchdringungstest (Hohlkoerper-Pruefkoerper) Hier im Shop bestellen
- Demineralisiertes/destilliertes Wasser als Speise- und Schlussspuelwasser Hier im Shop bestellen
- Pflege-/Schmiermittel fuer Gelenkinstrumente (sterilisierbar, herstellerfreigegeben) Hier im Shop bestellen
- Restproteintest/Reinigungsindikatoren zur Kontrolle des Reinigungserfolgs (v. a. bei manueller Aufbereitung)
- Persoenliche Schutzausruestung: fluessigkeitsdichte Einmalhandschuhe, Schutzbrille, Schutzkleidung
Zubehör
- Beschriftbare Etiketten/Stift bzw. Etikettendrucker fuer Inhalts-, Chargen- und Verfallskennzeichnung Hier im Shop bestellen Digital mit ClavioSoft erstellen
- Instrumentensiebe, Trays und Lagerungshilfen passend zur validierten Referenzbeladung Hier im Shop bestellen
- Sortier-/Transportbehaelter mit Deckel fuer Sammeln und Transport (rein/unrein getrennt) Hier im Shop bestellen
- Vorlagen/Formulare: Risikobewertungs-Liste, Standardarbeitsanweisung 'kritisch B', Muster-Siebliste, Beladungsmuster, Chargenfreigabe-Beleg Digital mit ClavioSoft erstellen
- Lupe bzw. Lupenleuchte fuer die Sichtkontrolle auf Sauberkeit und Unversehrtheit
- Getrennte und gekennzeichnete Arbeitsflaechen fuer unreinen und reinen Bereich
- Aktuelle Herstellerangaben zur Aufbereitung (DIN EN ISO 17664) je Instrument als abrufbare Referenz
Quellen & Rechtsgrundlagen
- Infektionsschutzgesetz (IfSG).
- Verordnung (EU) 2017/745 über Medizinprodukte (MDR) sowie Medizinprodukterecht-Durchführungsgesetz (MPDG).
- Medizinprodukte-Betreiberverordnung (MPBetreibV), insb. § 8 „Aufbereitung von Medizinprodukten“ (Vermutungswirkung Abs. 2).
- KRINKO/BfArM: „Anforderungen an die Hygiene bei der Aufbereitung von Medizinprodukten“, Bundesgesundheitsbl. 2012; 55:1244–1310 – insb. Abschn. 1.2.1 (Risikobewertung & Einstufung) und 2.2 (Durchführung); Anlage 6 (Sachkenntnis des Personals).
- DIN EN ISO 17664 (Herstellerangaben zur Aufbereitung); DIN EN 13060 (Dampf-Kleinsterilisatoren, Klasse B).
Dieser Beitrag fasst die genannten Rechts- und Normquellen praxisnah zusammen und ersetzt nicht das Studium der Originaldokumente. Im Zweifel gelten die jeweils aktuellen Fassungen der Gesetze, Verordnungen und Empfehlungen.
Wie audit-ready ist Ihre Praxis?
Finden Sie in wenigen Minuten heraus, wie gut Sie auf die nächste Begehung vorbereitet sind — Kategorie für Kategorie, mit klarem Fortschritt und konkreten offenen Punkten. Komplett kostenlos mit ClavioSoft.
.png "Preview: Clavio DokuSoft")



Häufige Fragen zur Instrumentenaufbereitung nach RKI
Die wichtigsten Fragen und fachlich fundierten Antworten zur Instrumentenaufbereitung nach RKI – auf Basis von RKI/KRINKO und den einschlägigen Normen.